Return to FAQ Table of Contents
The exact definition of "gene" depends on the context. In the context of genome annotation, a gene has at least a name and is defined by a collection of related RNA transcript sequences ("isoforms"). The naming of genes and the assignment of the most important transcript sequences is often done manually by a group of biological literature curators. For human, genes names are created by the Human Gene Nomenclature Committee (HGNC, formerly HUGO). Non-human species have similar annotation groups, e.g. Mouse Genome Informatics, Wormbase, Flybase, etc.
Transcripts are defined as RNA molecules that are made from a DNA template. Databases like the ones at the National Library of Medicine's NCBI or the European Bioinformatics Institute (EBI) collect these transcript sequences from biologists working on a gene. Every transcript has a unique identifier (accession), a gene that it is assigned to, a sequence, and a list of exon chrom/start/end coordinates on a chromosome. Usually every transcript is assigned to only a single gene. In the Genome Browser, transcript tracks often end with the word "Genes", e.g. "Ensembl Genes", "NCBI RefSeq Genes" or "UCSC Genes", but they really represent transcripts on chromosomes of a genome assembly.
For example, using the databases by NCBI, the gene with the gene symbol BRCA1 has 5 protein-coding transcripts or isoforms. The first transcript has the NCBI accession number NM_007294.3 which produces the protein with the accession NP_009225.1. In the human genome, it is located on chromosome 17, where it is comprised of 23 exons. On the version hg38/GRCh38 of the human genome, these exons cover the DNA nucleotides 43044295 to 43125483.
Gene symbols like BRCA1 are easy to remember but sometimes change and are not specific to an organism. Therefore most databases internally use unique identifiers to refer to sequences and some journals require authors to use these in manuscripts.
The most common accession numbers encountered by users are either from Ensembl, GENCODE or RefSeq. Human Ensembl/GENCODE gene accession numbers start with ENSG followed by a number and version number separated by a dot, e.g. "ENSG00000012048.21" for latest BRCA1. Every ENSG-gene has at least one transcript assigned to it. The transcript identifiers start with with ENST and are likewise followed by a version number, e.g. "ENST00000619216.1". Additional details on Ensembl IDs can be found on the Ensembl FAQ page.
NCBI refers to genes with plain numbers, e.g. 672 for BRCA1. Manually curated RefSeq transcript identifiers start with NM_ (coding) or NR_ (non-coding), followed by a number and version number separated by a dot, e.g. "NR_046018.2". If the transcript was predicted by the NCBI Gnomon software, the prefix is XM_ but these are rare in human. A table of these and other RefSeq prefixes can be found on the NCBI website.
Researchers sequence cDNA sequences and send these to NCBI Genbank. The Genome Browser shows these sequences in the Genbank or the EST track (if the cDNA is just a single read from the 5' or 3' end). From the alignment of the cDNAs and ESTs, the NCBI RefSeq group manually creates a smaller set of representative transcripts which we display as the RefSeq Curated track. Automated programs like UCSC's or Ensembl's gene build software do the same, just in software, which is more systematic but also more error-prone. With the arrival of GENCODE, Ensembl added a manual curation to their human and mouse transcripts. NCBI has added an automated prediction software (Gnomon) which we show in the "RefSeq Predicted" track.
There are many other tracks in the group "Genes and Gene Predictions". Genscan and N-Scan are older transcript predictor algorithms that are based on the genome sequence alone. Augustus and AceView are automated gene-predictors that use cDNA and EST data. These and similar gene tracks are only relevant when you are working on a particular locus where you think that the manually curated gene models (Ensembl and RefSeq) have errors.
To illustrate differences between the most common gene tracks, here is an overview of a few different tracks on human (hg38) and how many transcripts they contain as of March 2019:
| Track name | Number of transcripts |
|---|---|
| Known Gene (Gencode comprehensive V29) | 226,811 |
| Known Gene (Gencode basic V29) | 112,634 |
| NCBI RefSeq Predicted Transcripts | 94,389 |
| UCSC RefSeq (Curated) | 80,694 |
| NCBI RefSeq Curated | 73,080 |
| CCDS | 32,506 |
Officially, the Ensembl and GENCODE gene models are the same. On the latest human and mouse genome assemblies (hg38 and mm10), the identifiers, transcript sequences, and exon coordinates are almost identical between equivalent Ensembl and GENCODE versions (excluding alternative sequences or fix sequences).
GENCODE uses the UCSC convention of prefixing chromosome names with "chr", e.g. "chr1" and "chrM", but Ensembl calls these "1" or "MT". At the time of writing (Ensembl 89), a few transcripts differ due to conversion issues. In addition, around 160 PAR genes are duplicated in GENCODE but only once in Ensembl. The differences affect fewer than 1% of the transcripts. Apart from gene annotation itself, the links to external databases differ.
The GENCODE Release History shows the release dates and can be linked to corresponding Ensembl releases. You can download the gene transcript models from the website https://gencodegenes.org or from http://ensembl.org. For most applications, the files distributed on the GENCODE website should be easier to use, as the third party database links are easier to parse and the sequence identifiers match the UCSC genome files, at least for the primary chromosomes.
Additional information on this question can be found on the GENCODE FAQ page.
Different institutions have different rules on how they annotate genes. E.g. RefSeq's criteria are more stringent, so there are fewer RefSeq transcripts than Ensembl/GENCODE transcripts. Also, RefSeq transcripts have their own sequences independent of the genome assembly, so certain population-specific variants may be in RefSeq that are entirely missing from the reference genome sequence. This has the important implication that the position of genome variants are harder to map to RefSeq transcripts than for GENCODE since RefSeq transcripts can have additional sequence or missing sequence relative to the genome.
The links from either transcript model to other gene-related databases are different. In general, it seems that high-throughput sequencing data results, e.g. RNA-seq, are often using Ensembl/GENCODE annotations and human genetics results are reported using RefSeq annotations. It depends on your particular project which gene model set you want to use. Over time, the two transcript databases have been and are becoming more similar.
The "UCSC Genes" track, also called "Known Genes", is available only on assemblies before hg38. It was built with a gene predictor developed at UCSC. This gene predictor uses protein, EST and cDNA annotations to derive a relatively restricted gene transcript set. The software is no longer in use and there are no plans to release the track on newer human assemblies. It was last used for the mm10 mouse assembly. We are considering updating the hg19 annotation produced by this software and are interested in any user feedback on the topic.
The "GENCODE Gene Annotation" track contains data from all versions of GENCODE. "Ensembl Genes" track contains just a single Ensembl version. See the previous question for the differences between Ensembl and GENCODE.
"GENCODE" is the default gene track on hg38 (similar to "Known Genes" on hg19), which means that it is associated with a large amount of third party information when you click on a gene. This related information is also available using the Table Browser. This GENCODE track is updated periodically to match the latest GENCODE release. "All GENCODE" is a super-track that contains all versions of GENCODE as sub-tracks, but these tracks have less third-party information. Sub-tracks are never removed from "All GENCODE", and new sub-tracks are added as there are additional GENCODE releases.
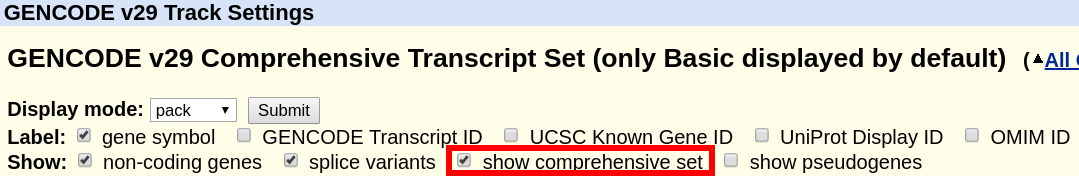
The "GENCODE" track offers a "basic" gene set, and a "comprehensive" gene set. The "basic" gene set represents a subset of transcripts that GENCODE believes will be useful to the majority of users. The "basic" gene set is defined as follows in the GENCODE FAQ:
"Identifies a subset of representative transcripts for each gene; prioritises full-length protein coding transcripts over partial or non-protein coding transcripts within the same gene, and intends to highlight those transcripts that will be useful to the majority of users."
A more comprehensive definition can also be found in the Esnembl FAQ. By default, the track displays only the "basic" set. In order to display the complete "comprehensive" set, the box can be ticked at the top of the GENCODE track description page.
RefSeq gene transcripts, unlike GENCODE/Ensembl/UCSC Genes, are sequences that can differ from the genome. They need to be aligned to the genome to create transcript models. Traditionally, UCSC has aligned RefSeq with BLAT (UCSC RefSeq sub-track) and NCBI has aligned with splign. The advantages of the UCSC alignments are that they are updated more frequently and are available for older assemblies (like GRCh37/hg19), but they are less stable and they are not the official alignments. Therefore we recommend working with the NCBI annotations. When an assembly has an "NCBI RefSeq" track, we show it by default and hide the "UCSC RefSeq" track.
In some rare cases, the NCBI and UCSC exon boundaries differ. Activating both RefSeq and UCSC RefSeq tracks helps you investigate the differences. Activating the RefSeq Alignments track shows NCBI's splign alignments in more detail, including double lines where both transcript and genomic sequence are skipped in the alignment. When available, the RefSeq Diffs subtrack may be helpful too. The upcoming MANE gene set will contain a set of high-quality transcripts that are 100% alignable to the genome and are part of both RefSeq and Ensembl/GENCODE but at the time of writing this project is at an early stage.
An anecdotal and rare example is SHANK2 and SHANK3 in hg19. It is impossible for either NCBI or BLAT to get the correct alignment and gene model because the genome sequence is missing for part of the gene. NCBI and BLAT find slightly different exon boundaries at the edge of the problematic region. NCBI's aligner tries very hard to find exons that align to any transcript sequence, so it calls a few small dubious "exons" in the affected genomic region. GENCODE V19 also used an aligner that tried very hard to find exons, but it found small dubious "exons" in different places than NCBI. The RefSeq Alignments subtrack makes the problematic region very clear with double lines indicating unalignable transcript sequence.
When reporting results as RefSeq coordinates, e.g. as HGVS, in research articles, please specify the RefSeq annotation release and also the RefSeq transcript ID with version (e.g. NM_012309.4 not NM_012309). Different RefSeq transcript versions have different sequence (for example, more sequence may be added to the UTRs or even the CDS), and so the transcript coordinates often change from one version to the next.
The Consensus Coding Sequence Project is a list of transcript coding sequence (CDS) genomic regions that are identically annotated by RefSeq and Ensembl/GENCODE. CCDS undergoes extensive manual review and you can consider these a subset of either gene track, filtered for high quality. The CCDS identifiers are very stable and allow you to link easily between the different databases. As the name implies, it does not cover UTR regions or non-coding transcripts.
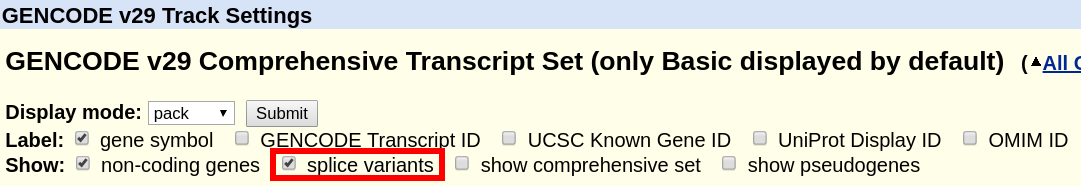
For the tracks "UCSC Genes" (hg19) or "GENCODE Genes" (hg38), click on their title and on the configuration page, uncheck the box "Show splice variants". Only a single transcript will be shown. The method for how this transcript is selected is described in the track documentation below the configuration settings.
For the track NCBI RefSeq (hg38), you can activate the subtrack "RefSeq HGMD". It contains only the transcripts that are part of the Human Gene Mutation Database.
We have data tables named knownCanonical available for different assemblies comprised of a single transcript/isoform per gene.
For hg19, the knownCanonical table is a subset of the UCSC Genes track. It was generated by identifying a canonical isoform for each cluster ID, or gene. Generally, this is the longest isoform. It can be downloaded directly from the hg19 downloads database or by using the Table Browser.
For hg38, the knownCanonical table is a subset of the GENCODE v29 track. As opposed to the hg19 equivalent which generally used the longest isoform for indentification, this table is defined as follows:
knownCanonical identifies the canonical isoform of each cluster ID or gene using the ENSEMBL gene IDs to define each cluster. The canonical transcript is chosen using the APPRIS principal transcript when available. If no APPRIS tag exists for any transcript associated with the cluster, then a transcript in the BASIC set is chosen. If no BASIC transcript exists, then the longest isoform is used.
It can be downloaded directly from the hg38 downloads database or by using the Table Browser.
For automated analysis, if you are doing NGS analysis and you need to capture all possible transcripts, GENCODE provides a comprehensive gene set. For human genetics or variant annotation, a more restricted transcript set is usually sufficient and "NCBI RefSeq" is the standard. If you are only interested in protein-coding annotations, CCDS or UniProt may be an option, but this is rather unusual.
For manual inspection of exon boundaries of a single gene, and especially if it is a transcript that is repetitive or hard to align (e.g. very small exons), look at the UCSC RefSeq track and watch for differences between the NCBI and UCSC exon placement. You can also BLAT the transcript sequence. Manually look at ESTs, mRNAs, TransMap and possibly Augustus, Genscan, SIB, SGP or GeneId in obscure cases where you are looking for hints on what an alternative splicing could look like.
You may also find the Gene Support public session helpful. This session is a collection of tracks centered around supporting evidence for genes.
[an error occurred while processing this directive]