[an error occurred while processing this directive]
barChart and bigBarChart Track Format
The barChart (and bigBarChart) track format displays a graph of category-specific values over
genomic regions, similar to the
GTEx Gene track.
This format is useful for displaying gene expression and other datasets where it is desirable to
compare a set of variables over genomic regions.
While a barChart track can effectively show datasets with single values for each
variable (e.g. comparing individual samples),
the format provides specific features to display studies comprised of a large set
of samples for each variable (e.g. comparing tissues with multiple samples for each tissue).
In this usage, the main genome browser display presents a graph of summary values (e.g. medians)
for each variable, and the distribution of sample values across variables is
shown via a boxplot graph shown on the details page for each region.
The barChart format is available as a standalone plain text bed6+ format
for use with smaller datasets as a custom track, and as a binary indexed format (bigBarChart)
suitable for track hubs and custom tracks.
The bigBarChart format provides more track customization
features (i.e. schema customization, and label configuration support), and
is recommended for users who can use command-line tools and have web-accessible data storage. If
you do not have web-accessible data storage, please see the
Hosting section of the Track Hub Help documentation.
barChart format files are converted to bigBarChart files using the program
bedToBigBed, run with the -as option to pull in a special
autoSql (.as)
schema file defining the fields of the bigBarChart.
barChart format definition
The following autoSql definition illustrates the basic schema supporting barChart (and
bigBarChart) tracks.
table bigBarChart
"bigBarChart bar graph display"
(
string chrom; "Reference sequence chromosome or scaffold"
uint chromStart; "Start position in chromosome"
uint chromEnd; "End position in chromosome"
string name; "Name or ID of item"
uint score; "Score (0-1000)"
char[1] strand; "'+','-' or '.'. Indicates whether the query aligns to the + or - strand on the reference"
string name2; "Alternate name of item"
uint expCount; "Number of bar graphs in display, must be <= 100"
float[expCount] expScores; "Comma separated list of category values."
bigint _dataOffset; "Offset of sample data in data matrix file, for boxplot on details page, optional only for barChart format"
int _dataLen; "Length of sample data row in data matrix file, optional only for barChart format"
)
Column Explanations
The first 6 fields of the barChart format are the same as the first 6 fields of the standard
BED format. The name2 field provides an alternate
item name, useful if you would like to associate multiple transcripts to a single gene locus,
different variables to the same experiment type, etc. The expCount and expScores fields are
used as in the Microarray format; they define the
number of categories and a value for each category (see example #1 below).
The _dataOffset and _dataLen fields are used internally by the track to locate sample values
for a region in an optional matrix file containing all sample values.
These values are used to draw a boxplot of all sample data on the details page for the bar chart.
When a matrix file is not supplied, these fields should be set to 0.
(As a convenience, these fields are optional for barChart custom tracks).
When creating bigBarChart files, we encourage you to customize the title and field
descriptions of the prototype autoSql schema to better describe your data.
In the example below, the name field of the track refers to a transcript, while the name2
field represents a gene:
table xyzGeneExpression
"XYZ gene expression barChart"
(
string chrom; "Reference sequence chromosome or scaffold"
uint chromStart; "Start position in chromosome"
uint chromEnd; "End position in chromosome"
string name; "Transcript name"
uint score; "Score (0-1000), derived from total expScores (below)"
char[1] strand; "+, -, or ., indicating orientation of the item"
string name2; "Gene name"
uint expCount; "Number of tissues"
float[expCount] expScores; "Comma separated list of median expression in RPKM for each tissue."
bigint _dataOffset; "Offset of sample data in data matrix file"
int _dataLen; "Length of sample data row in data matrix file"
)
Customing this file will make your data more easily interpreted by users, who will
see the field descriptions when accessing the track data from
the Table Browser,
when viewing items on the Genome Browser details
pages (via the "view table schema" link),
and (for users who download files), from the -as option of the bigBedInfo tool.
Creating barChart and bigBarChart custom tracks
The steps for creating barChart tracks differ from the process for creating
bigBarChart tracks. The steps also differ based on whether you have an input
matrix file (generated perhaps from an RNA-Seq differential expression analysis pipeline) or not.
If you have an expression matrix-like file, skip to Example #3, otherwise
follow example 1 below.
Example #1
In this example, you will create a barChart custom track using example bed6+3 data.
- Paste the following track line into the
custom track management
page for the human assembly hg38.
track type=barChart name="barChart Example One" description="A barChart file" barChartBars="adiposeSubcut breastMamTissue colonTransverse muscleSkeletal wholeBlood" visibility=pack
browser position chr14:95,081,796-95,436,280
# chrom chromStart chromEnd string name score strand name2 expCount expScores _dataOffset _dataLen
chr14 95086227 95158010 DICER1 999 - ENSG00000100697.10 5 2.94,11.60,38.00,6.69,4.89
chr14 95181939 95319906 CLMN 999 - ENSG00000165959.7 5 7.08,69.53,9.32,1.38,1.68
chr14 95417493 95475836 SYNE3 999 - ENSG00000176438.8 5 7.29,3.73,0.74,20.35,1.39
- Click the "submit" button.
After the file loads in the Genome Browser, you should see an automatically colored bar
graph with 5 bars. Hovering the mouse over any of the individual bars will display the name of the
particular bar ("wholeBlood", "adiposeSubcut", ...) as well as the value associated with that bar
(10.94, 0.74, ...). The order of bar names in the barChartBars field of track line should exactly
match the order of the values in the expScores field.
Example #2
In this example, you will create a bigBarChart track out of an existing bigBarChart format file,
located on the UCSC Genome Browser http server. This file contains data for the hg38 assembly.
To create a custom track using this file:
- Construct a track line referencing the file:
track type=bigBarChart name="bigBarChart Example One" description="A bigBarChart file" barChartBars="adiposeSubcut breastMamTissue colonTransverse muscleSkeletal wholeBlood" visibility=pack bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/barChart/hg38.gtexTranscripts.bb
browser position chr14:95,081,796-95,436,280
- Paste the track line into the custom track management
page for the human assembly hg38.
- Click the "submit" button.
After the file loads in the Genome Browser, you should see an automatically colored bar
graph with 5 bars. The same rules apply to bigBarChart custom tracks as barChart custom tracks
in that the order of names in the barChartBars field should exactly match the order of values
from the expScores field in the bigBarChart file.
Example #3
In this example, you will use the helper scripts expMatrixToBarchartBed and bedJoinTabOffset
on example matrix and category files in order to generate a bed6+5 barChart format file, which
can be loaded as a custom track into the Genome Browser.
The matrix file is a tab-separated (must be tabs, not spaces) file of the following form, perhaps
resulting from an RNA-Seq analysis pipeline. Please note that the first line must describe each
column like in the example snippet below.
transcript sample1 sample2 sample3 sample4 sample5 ...
transcriptName value1 value2 value3 value4 value5 ...
The categories file then provides more meta information about this matrix file. It is a two column,
tab-separated file that maps the samples in the matrix file to a specific category:
sample1 category1
sample2 category1
... ...
sampleA category2
sampleB category2
... ...
sampleX category3
sampleY category3
... ...
Each column in the first line of the matrix file must be found in the categories file.
We have provided an example category file
and matrix file to follow
along with the rest of this example.
To create a custom track in this form, follow the below steps:
- Create a bed6+1 file to use as a map for the items in your matrix (does not need to be
all-inclusive). This file, with example lines like:
chr14 95086227 95158010 ENSG00000100697.10 999 - DICER1
will be one
of three input files to the helper script expMatrixToBarchartBed. For an example file to
follow along with the rest of this example, you can download an example bed6+1 file
here.
- Download the helper programs expMatrixToBarchartBed and bedJoinTabOffset
from the utilities directory
appropriate for your operating system.
- Make sure the programs downloaded above are in your system's PATH variable. For example,
if you downloaded the programs to your $HOME/Downloads directory, set your PATH variable
accordingly:
export PATH=$PATH:$HOME/Downloads
- Now run expMatrixToBarchartBed (which in turn runs bedJoinTabOffset) like so:
expMatrixToBarchartBed categoriesFile matrixFile bedInputFile outputBed
The argument outputBed will be a bed6+5 file, with the expCount, expScores, _dataOffset, and
_dataLen fields computed for you, for example:
chr14 95086227 95158010 ENSG00000100697.10 999 - DICER1 5 10.94,11.60,8.00,6.69,4.89 93153 26789
expMatrixToBarchartBed will also
output the order of the scores in the expScores field, which you can then
copy and paste into the barChartBars field of the custom track line so
the bars displayed in the browser match the right values:
The columns and order of the groups are:
#chr start end name score strand name2 expCount expScores;adiposeSubcut breastMamTissue colonTransverse muscleSkeletal wholeBlood _offset _lineLength
If you have already pre-computed expCount and
expScores, and just need offsets into your matrix file for a more descriptive details
page, run only bedJoinTabOffset like so:
bedJoinTabOffset matrixFile exampleBed6+3 outBed
- Now that we have a bed6+5 format file that corresponds to the barChart format, we can
construct a track line and prepend it to our bed6+5 file so the Genome Browser will
recognize it:
track type=barChart name="barChart Example" description="A barChart file" barChartBars="adiposeSubcut breastMamTissue colonTransverse muscleSkeletal wholeBlood" barChartMetric=median" visibility=pack
- Use the upload button on the custom track management
page for the human assembly hg38 to upload the file just created.
- Click the "submit" button.
expMatrixToBarchartBed automatically computes the median values for all the samples in
the matrix file, which is useful when your experiment contains data from 8000 samples (like
the GTEx data). Furthermore, expMatrixToBarchartBed can compute the mean value of all
samples in a category of the matrix file (instead of the default median), in addition to allowing
for a specific ordering of the expScores field. NOTE: Set the barChartMetric setting to 'mean' if
you use this option of expMatrixToBarchartBed. For more information about
expMatrixToBarchartBed or bedJoinTabOffset, run the program with no arguments
to get a usage message.
Example #4
In this example, you will use the bed6+5 file created in Example 3 to create a bigBarChart file,
allowing the data to be remotely accessed and exist within a track hub. The track settings for
bigBarChart on a hub can be viewed here.
- If not already completed, follow steps 1-4 from Example 3 above, or
download the example bed6+5 file here
- Download the fetchChromSizes and bedToBigBed programs from the
utilities directory appropriate to your
operating system.
- Use fetchChromSizes to create a chrom.sizes file for the UCSC database you
are working with (hg38 for these examples). Alternatively, you can download the
chrom.sizes file for any assembly hosted at UCSC from
our downloads page (click on "Full
data set" for any assembly). For example, the hg38.chrom.sizes file for the hg38
database is located at
http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/hg38.chrom.sizes.
- Save the autoSql file barChartBed.as to your computer.
- Run bedToBigBed to create the bigBarChart file:
bedToBigBed -as=barChartBed.as -type=bed6+5 inputBed hg38.chrom.sizes output.bigBed
- Move the newly constructed bigBarChart file to a web accessible http, https, or ftp location.
- Construct a custom track line with a bigDataUrl parameter pointing to the newly created
bigBarChart file. If the matrix and category files used to make the precursor barChart file
are also moved to an http, https, or ftp location, we can point to them on the custom
track line as well (all settings must be on the same line):
track type=bigBarChart name="bigBarChart Example One" description="A bigBarChart file"
barChartBars="adiposeSubcut breastMamTissue colonTransverse muscleSkeletal wholeBlood"
barChartMetric=median barChartUnit=RPKM
bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/barChart/hg38.gtexTranscripts.bb
barChartMatrixUrl=http://genome.ucsc.edu/goldenPath/help/examples/barChart/exampleMatrix.txt
barChartSampleUrl=http://genome.ucsc.edu/goldenPath/help/examples/barChart/exampleSampleData.txt
visibility=pack
- To fully take advantage of creating a bigBarChart file and the barChartMatrixUrl and
barChartSampleUrl supporting files, create a Track Hub and
use a stanza such as the following:
track exampleBarChartTrack
type bigBarChart
visibility full
shortLabel exBarChart
longLabel Simple example bar chart track
barChartBars adiposeSubcut breastMamTissue colonTransverse muscleSkeletal wholeBlood
barChartColors #FF6600 #33CCCC #CC9955 #AAAAFF #FF00BB
barChartLabel Tissues
barChartMetric median
barChartUnit RPKM
bigDataUrl http://genome.ucsc.edu/goldenPath/help/examples/barChart/hg38.gtexTranscripts.bb
barChartMatrixUrl http://genome.ucsc.edu/goldenPath/help/examples/barChart/exampleMatrix.txt
barChartSampleUrl http://genome.ucsc.edu/goldenPath/help/examples/barChart/exampleSampleData.txt
Please note, the fields in your barChartBars line must match the terms
in your categories file (exampleBarChartSamples.txt) in order for the boxplot display to show
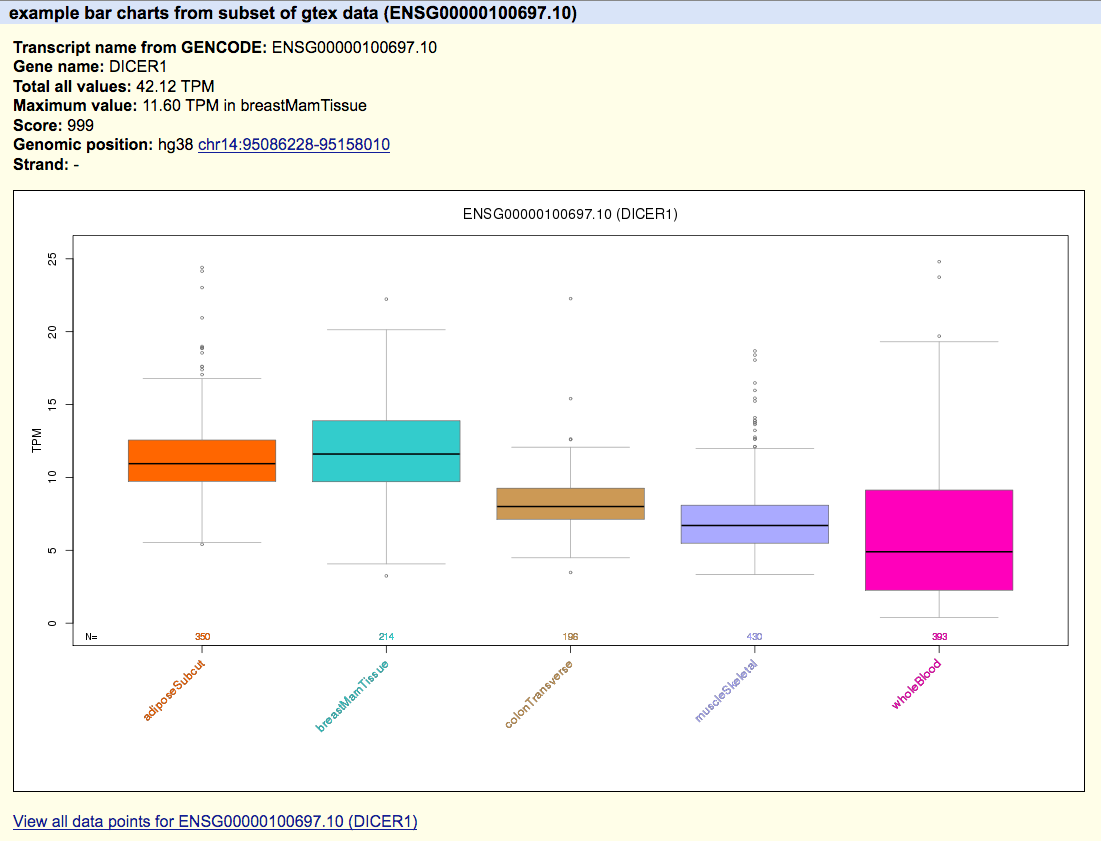
up on the details page for tracks. Below is an example image indicating the benefit of using
these files in a hub, note the "View all data points for..." link that allows extracting data
from the matrix file (exampleBarChartMatrix.txt) specific for this named item.
Sharing your data with others
If you would like to share your barChart/bigBarChart data track with a colleague, learn how to create a URL by
looking at Example 11 on this page.
Extracting data from the bigBarChart format
Because bigBarChart files are an extension of bigBed files, which are indexed binary files, it can
be difficult to extract data from them. UCSC has developed the following programs to assist
in working with bigBed formats, available from the
binary utilities directory.
-
bigBedToBed — converts a bigBed file to ASCII BED format.
-
bigBedSummary — extracts summary information from a bigBed file.
-
bigBedInfo — prints out information about a bigBed file. Use the -as option to see the file field descriptions.
As with all UCSC Genome Browser programs, simply type the program name (with no parameters) at the
command line to view the usage statement.
Troubleshooting
If you encounter an error when you run the bedToBigBed program, check your input
file for data coordinates that extend past the end of the chromosome. If these are present, run
the bedClip program
(available here) to remove the problematic
row(s) in your input file before running the bedToBigBed program.
[an error occurred while processing this directive]